Enabling Valence Delocalization in Iron(III) Macrocyclic Complexes through Ring Unsaturation
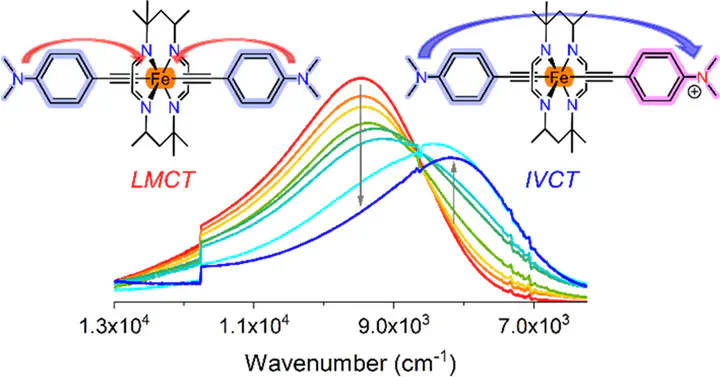 Image credit: Unsplash
Image credit: Unsplash
Abstract
The complexes [FeIII(HMC)(C2DMA)2]CF3SO3 ([2]OTf) and [FeIII(HMTI)(C2Y)2]CF3SO3 ([3a–c]OTf) have been prepared and thoroughly characterized (HMC = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane; HMTI = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-1,3,8,10-tetraene; Y = Fc (ferrocenyl, [3a]OTf), 4-(N,N-dimethyl)anilino (DMA, [3b]OTf), or 4-(N,N-bis(4-methoxyphenyl)anilino (TPA, [3c]OTf); OTf– = CF3SO3–)). Vibrational and electronic absorption spectroelectrochemical analyses following one-electron oxidation of the ethynyl substituent Y revealed evidence of strong coupling in the resultant mixed valent species for all HMTI-based complexes. However, the analogous mixed valent ion based on [2]OTf appeared to be more localized. Thus, the tetra-imino macrocycle HMTI has enabled significant valence delocalization along the −C2–FeIII–C2– bridge. Electron paramagnetic resonance and Mössbauer spectroscopic studies of [3b]OTf reveal that the π-acidity of HMTI lowers the energy of the FeIII dπ orbitals compared to the purely σ-donating HMC. This observation provides a basis for the interpretation of the macrocycle-dependent valence (de)localization.